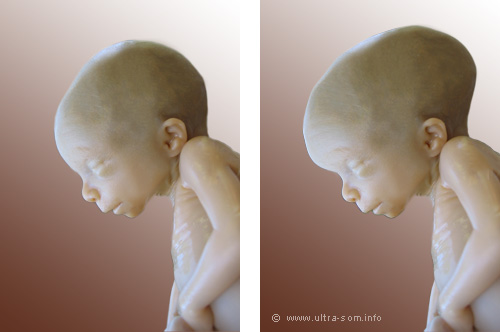
Por definición Hidrocefalia Congénita "Neonatal o Perinatal" se desarrolla lntrauterinamente durante el embarazo, pero puede detectarse hasta el parto o el período de recién nacido, aunque muchas veces.
las manifestaciones clínicas evolutivas se presentan en períodos posteriores haciendo díficil diferenciar entre una causa congénita y otra adquirida, sin embargo el carácter congénito se define por alteraciones patológicas del tubo neural durante la tercera y cuarta semana de vida embrionaria. Se trata de un estado patológico de diferentes causas, donde hay un incremento de la cantidad de líquido cefalorraquídeo (LCR) ventricular, por una ruptura de equilibrio entre la formación y absorción del mismo, que lleva a un aumento de la presión intracrancal con disminución concomitante de la sustancia cerebral, sin que esto deba asociarse siempre a macrocefalia.
En relación al sexo hay muy pocas variaciones, pero pareciera que los varones son más afectados por hidrocefalia sola, posiblemente debido a una asociación génetica ligada al cromosoma "X".
CLASIFICACION:
- Se distingue dos grandes grupos: La hidrocefalia no comunicante y comunicante.
I) Hidrocefalia no comunicante: Hay una interferencia en la circulación normal de L.C.R. dentro del sistema ventricular desde uno o ambos agujeros de Monro, Acueducto de Silvio, IV ventrículo, Luschka y Magendie.
II) Hidrocefalia Comunicante: Hay bloqueo de la absorcíón de L.R.C. en las vellosidades aracnoideas.
ETIOLOGIA:
Las causas pueden ser prenatales, genéticas o familiares que se asocia a anomalías cromosómicas, siendo estos factores responsables desconocidos para la mayoría de los autores, infecciosas y/o
parasitarias y las de causa posnatal que son los procesos expansivos e infecciosos.
I ) Hidrocefalia no comunicante
1) Lesiones Congénitas :
A) Obstruccion o estenosis de A. de Silvio
a) Gliosis
b) Forki n
c) Estenosis verdadera
d) Septum '
B) Atresia del foramen de Luschka y Magendie (Dandy Walker)
C) Masas:
a) Quistes intracraneanos Benignos
b) M.A.V.
c) Tumores
2) Lesiones Adquiridas
A) Estenosis de acueductos de Silvio (Gliosis)
B) Inflamaciones ventriculares y cicatrices
C) Masas:
a) Tumores
b) Masa no neoplastica
D) Craneo
a) Platisbasia
b) Acondroplasia
3) Hidrocefalia Comunicante
1 . Lesiones Congénitas
a)Malformaciones de Arnold Chiari
b) Encefalocele
c) Inflamacion de Leptomeninges
d) Lisencefalia
e) Ausencia congénita de vellocidades aracnoideas '
2 . Lesiones Adquiridas
A) Inflamaciones de Leptomeninges :
a) Infecciones
b) Hemorragias
c) Cuerpos extranos
B) Masas
a) Tumores
b) No neoplasticas
3 . Hipersecresion de LC. R .
a ) Papilomas de plexo coroideo
4 . Miscelanea
a) Deficiencia vitamínica: complejo B
b) Toxinas en período embrionario
c) Drogas: Esteroides, vitamina A, AC. Valpróico y otros .
d) Genéticas
5. Idiopaticas
DIAGNÓSTICO:
El diagnóstico prenatal de las hidrocefalias congénitas es posible incluso en la 13a semana de gestación. Posteriormente el aumento del perímetro cefálico, evaluación del fondo de ojo, la radiografía simple de cráneo, la ecografía transfontanelar, tomografía axial computariza (TAC) de cráneo y la Resonancia magnética son utilizadas para su diagnóstico.
Las manifestaciones clínicas dependen de la edad de la instalación del desequilibrio entre producción y absorción del L.C.R. El crecimiento anormal del tamaño cefálico ocurre invariablemente en la hidrocefalia congénita y en la de instauración durante la lactancia.
En los recién nacidos puede haber solo irritabilidad psicomotora, hiporexia marcada con alteraciones en
la succión, insuficiencia postnatal o presentar signos severos de paro cardiorespiratorio, coma o herniación transtentorial por hipertensión intracreana severa. Los signos de parinaud y mac-ewen aparecen en forma más posterior.
TRATAMIENTO:
El mejor tratamiento es la inserción de una válvula, que no es la cura de la hidrocefalia ya que el daño al
tejido cerebral permanece y con el uso de una válvula de derivación ventrículo peritoneal (DVP) se logra mantener la presión bajo control por medio del drenaje del líquido cefalorraquídeo excesivo. Un tratamiento alternativo podría ser la ventriculostomia evitando la necesidad de una válvula sin embargo no se puede tratar todos los tipos de hidrocefalia con este método.
COMPLICACIONES:
Las complicaciones en los sistemas de derivación son por fallas mecánicas e infecciosas. El pronóstico
depende del diagnóstico temprano y el éxito del tratamiento reduce el daño cerebral y preserva la vida
del paciente.
REFERENCIAS